What's the process behind tuning our analytes to the mass spectrometer’s optics? And why is that so important?
Previously, I spoke about how to focus our mass spectrometer’s source to maximize our analytes signal. We were interested in the detection of naloxone, buprenorphine, norfentanyl, and methadone in urine and determining their source parameters.
If you recall, I mentioned that most of the MS parameters and transitions are readily available for our analytes from multiple sources. This of course allows one to “plug-n-chug” the values. However, this step doesn’t really serve us well because we need to determine the values that our mass spectrometer responds to.
Let’s quickly recap:
- -we have our matrix and our analytes and a Shimadzu NexeraX2 LC with a 5500 Sciex MS
- -We did some searching and eventually settled on a poster we found on the Biotage website:
Sample Preparation Strategies for Urine Panels with 50 or More Drugs and Metabolites Analyzed by LC-MS/MS - -Next, we sought to determined what the best global values would be for temperature (TEM), curtain gas (CUR), ion spray voltage (IS), and our gas flow rates for GS1 and GS2 for all our analytes. We used the FIA feature of the Sciex 5500 mass spectrometer (Analyst) to assist us with this and found the following settings worked best for all our analytes:
- TEM: 600 °C
- CUR: 30
- IS: 1500 V
- GS1: 50
- GS2: 70
-
This step allowed us to maximize the number of droplets formed in the source, which increases the abundance of analytes entering the low vacuum stage of the mass spectrometer.
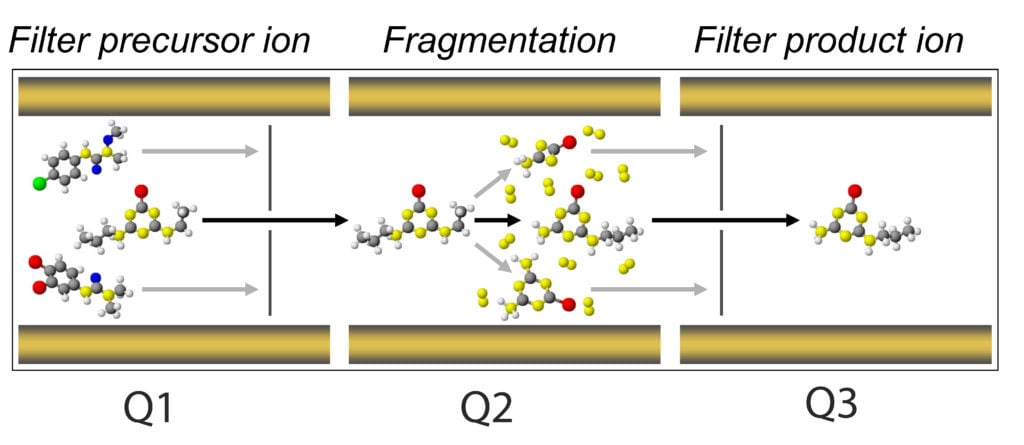
- Now we need to consider the second stage of our mass spectrometer, the optics. The optics, which are sometimes referred to as Q1 (the first quadrupole), Q2 (the collision chamber), and Q3 (the second quadrupole). Ultimately, what we want to achieve is establishing a qualifying fragmentation pattern for each of our analytes.
 What does this mean?
What does this mean?
It means each time an analyte makes its way into the optics, travels from Q1 to Q2 and to Q3, it will do so in a unique way that will positively identify this analyte and only this analyte.
We perform this function through a process call multiple reaction monitoring or MRM – other manufactures have similar names for this such as SRM.
The main principle behind a MRM is filtering your analyte in Q1, or the precursor ion, so that only precursors of a set m/z will pass through Q1. Upon entering Q2, a voltage is applied with an inert gas (typically nitrogen or argon) to induce the fragmentation of your precursor but in a predictable and reproducible manner.
This is where we tune for our collision energy (CE) parameter. At an appropriate CE, your precursor will yield daughter ions, aka product ions. The potential applied at the exit of Q2 is the collision cell potential (CXP). This potential is tuned to ensure successful ion acceleration out of Q2 and into Q3. The ions accelerated into Q3 are then filtered based on the selected product ions.
Each product ion possesses different intrinsic molecular connectivity, which is unique to its m/z, just like their precursor. However, multiple product ions can, and do, form in Q2. This places an opportunity for us where we can usually select two of the most stable product ions and use one for quantitative evaluation and the other as a qualitative metric.
That is, one precursor can yield multiple products, and in order to establish a unique MRM for precursor, we use on product ion for quantitation and second most stable product ion for qualitative identification.
Ultimately, we will have the retention time (from the LC) and two MRMs for one analyte MS, all of which are used for confirming the presence of a particular analyte.
Moving on:
Just as we tuned the source with FIA, we will do the same, however, with the source properties already established, and set up an infusion system. To do this, we make a cocktail of our analytes in methanol and draw them into a syringe.
To administer or infuse the tuning solution, you can either use the pump on the MS (it’s behind the door on the right-hand side in picture) or an alternate, like Harvard Apparatus. We plumb the tuning solution directly into the MS source, post HPLC flow. In this way, we don’t rely on chromatography, but solely the mass spectrometer’s ability to realize the analytes and the LC to provide the eluent.
Now that we have the physically set up the system to tune, we set up the software to do just that.
The parameters we will tune against for every analyte are the declustering potential (DP), collision energy (CE) and the collision cell exit potential (CXP):
- DP: the declustering potential. This parameter is specific for each analyte and is very important for maximizing our analytes’ entry into the MS optics. This potential ranges from 0 to 300 V in positive ion mode and -300 to 0 V for negative ion mode. This potential runs across the orifice, or entry point of the MS, and prevents ions from clustering with adducts (like sodium) or neutral species without causing analyte (or in-source) fragmentation.
- CE: the collision energy. This parameter controls the potential difference between Q0 and Q2 (collision cell). This is the amount of energy that the precursor ions receive as they are accelerated into the collision cell, where they collide with gas molecules and fragment and ranges from -300 to 300V.
- CXP: the exit cell potential. This parameter controls the potential difference between the focusing lenses and filter positioned at Q2. It is only used in MS/MS-type scans, where it transmits the ions into Q3.
With such a low number of analytes, it’s easier to manually tune your MS as opposed to autotune. But the choice is yours!
Once we collect these parameters, we’ll put them together to complete the MS portion of our method and next, we’ll move on to the LC.
For a good tutorial, go to Sciex.com and visit their Education section. They have a number of good tutorials on the basics of mass spectrometry as does LCGC’s CHROMacademy; however, the former requires registration and not all material is available without purchase. Nonetheless, they’re a good source for looking into the basics.
So, your MS source is tuned and ready to go? What about the quadrupoles? Want to know more about how to tune your MS optics to increase your analytes level of detection?